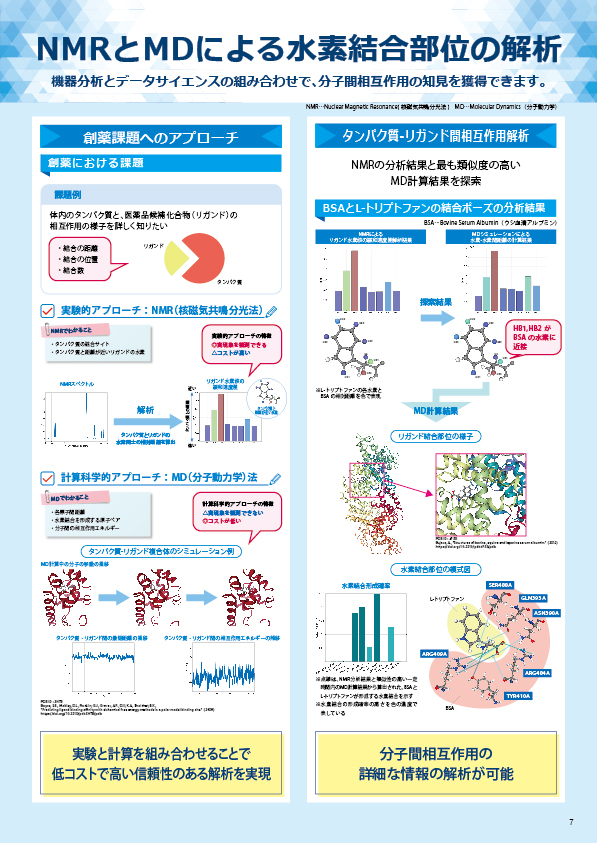
ミクロな原子構造を計算シミュレーションによって評価可能
概要
β-Ga2O3は広いバンドギャップを有し、優れた送電効率や低コスト化の面で次世代パワーデバイスや酸化物半導体の材料として期待されています。近年、β-Ga2O3はSiまたはSnのドーピングでn型化することが報告されています。本資料では、β-Ga2O3にSiもしくはSnをドープしたモデルに対して構造最適化計算を実施し、各ドーパントが結晶中でどのサイトを占有しやすいかを評価しました。続いて、得られた構造モデルから状態密度を計算し、ドーピングによる電子状態の変化を調査しました。
データ
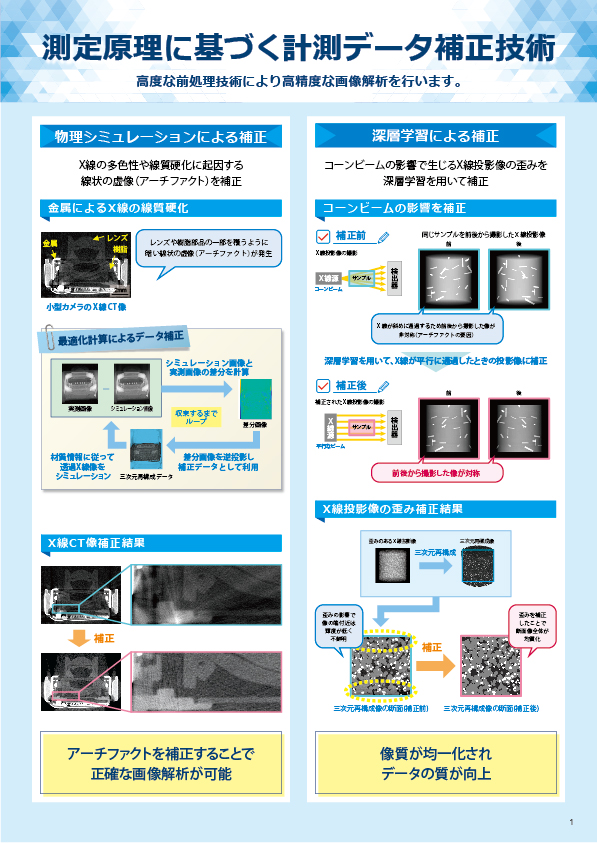
β-Ga2O3構造
β-Ga2O3の構造を下図※に示します。Gaの存在するサイトは、周囲のO原子が作る四面体の中心にあるサイト:Ga(1)と、周囲のO原子が作る八面体の中心にあるサイト:Ga(2)の2種類が存在します。

Si,Snドープ時の構造最適化
2種類存在するGaのサイトに対し、SiもしくはSnを置換したモデルを作成し、構造最適化計算を実施することで各ドーパント元素の存在サイトの同定を行いました。
構造最適化後の安定化エネルギー
| ドーパント | Si | Sn | ||
|---|---|---|---|---|
| サイト | Ga(1) | Ga(2) | Ga(1) | Ga(2) |
| エネルギー[eV/unit cell] | -22037.853 | -22037.667 | -22058.017 | -22058.224 |
- ドープされたSiはGa(1)サイト(4配位)の位置で安定化します。
- ドープされたSnはGa(2)サイト(6配位)の位置で安定化します。

構造最適化後のドーパント周囲の構造
| サイト | Ga(1) | Ga(2) | ||
|---|---|---|---|---|
| ドーパント | – | Si | – | Sn |
| 平均結合距離[Å] | 1.867 | 1.699 | 2.028 | 2.077 |
| 結合距離標準偏差 | 0.016 | 0.007 | 0.063 | 0.036 |
| 多面体体積[Å3] | 3.306 | 2.480 | 10.863 | 11.731 |
- SiはGa(1)サイトへ置換することで、四面体の歪みを小さくし、O原子との結合距離を短くします。
- SnはGa(2)サイトへ置換することで、八面体の歪みを小さくし、O原子との結合距離を長くします。
※図はVESTA(https://jp-minerals.org/vesta/jp/)で作成
状態密度
β-Ga2O3及びSi、Snドープβ-Ga2O3の状態密度(DOS)、部分状態密度(PDOS)を求めました。

計算結果
| ドーパント | – | Si | Sn |
|---|---|---|---|
| サイト | – | Ga(1) | Ga(2) |
| VBM[eV] | -0.8 | -5.2 | -5.1 |
| VBMを構成する主な軌道 | O 2p | O 2p | O 2p |
| CBM[eV] | 3.6 | -0.8 | -0.8 |
| CBMを構成する主な軌道 | Ga 4s | Ga 4s | Ga 4s Sn 5s |
| バンドギャップ[eV] | 4.4 | 4.4 | 4.3 |
| 光学ギャップ[eV] | 4.4 | 5.2 | 5.1 |
- Si、Snをドーパントとしたとき、いずれもフェルミ準位が伝導帯下端(CBM)に位置し、n型化が確認されました。
また、いずれのドーパントにおいても光学ギャップが広がることが確認されました。